Systems-level investigation of mucopolysaccharidosis IIIA identifies deficient synaptic activity as a key driver of disease progression
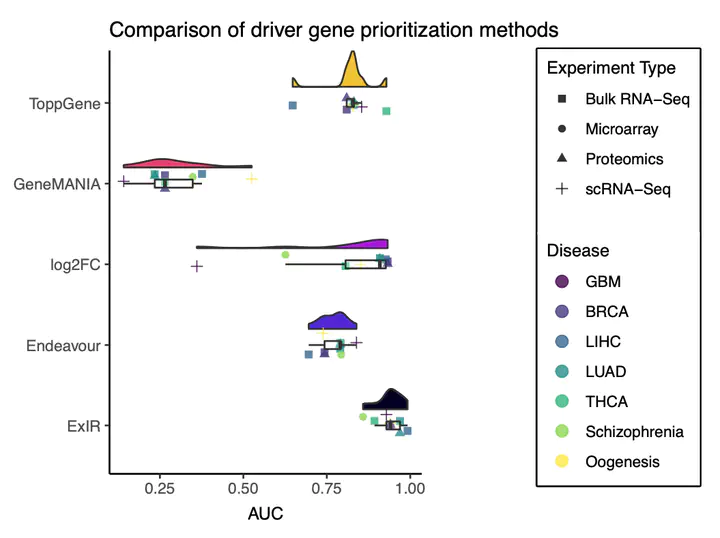 Image credit: Adrian Salavaty
Image credit: Adrian SalavatyAbstract
Mucopolysaccharidoses are lysosomal storage diseases that collectively represent a major cause of lethal, treatment-refractory childhood dementias 1–7 Clinically-useful interventions are hampered due to an incomplete understanding of their neuropathological origins. Using the zebrafish sgsh model of mucopolysaccharidosis IIIA 8 (MPS IIIA, Sanfilippo syndrome A), we conducted several ‘omics-based analyses, and developed and benchmarked a novel bioinformatic feature classification and ranking model for high-throughput datasets – ExIR – to prioritise important features in the progression of neurological manifestations of the disease. We find that the massive endolysosomal burden resulting from increased lysosomal storage of heparan sulfate and other secondarily accumulating substrates, such as sphingolipids, induces abnormal microtubule organisation and vesicle trafficking in neurons. This results in a gradual impairment of synaptic vesicle localisation at the presynaptic terminal and consequently impaired neuronal activity. Importantly, the endolysosomal phenotype in MPS IIIA zebrafish well-precedes the onset of neural pathology, though the larval MPS IIIA brain was found to be more susceptible to perturbation than wild type siblings. Collectively, these analyses demonstrate the presence of a progressive ‘functional neurodegenerative’ phenotype underpinning neurological disease in MPS IIIA. Our findings provide direct mechanistic evidence linking the well-described lysosomal storage basis for MPS IIIA to its disproportionately severe neural clinical involvement, enabling development and refinement of future therapeutic interventions for this currently untreatable disorder.
Key words
ExIR; Prioritization; Classification, Sanfilippo syndrome A; Zebrafish; Driver Genes; Biomarkers; Mediator Genes